WeCoNET Tutorial
This section of WeCoNET provides a detailed guide to navigate through the database. If you have any questions that are not covered on this page, feel free to send an email to Raghav Kataria.
Introduction
Wheat (Triticum aestivum), the most widely cultivated crop in the world, and counts to around 35% of the total grains
produced around the globe. In the recent years, the occurence of common bunt in wheat has affected the grain yield and quality of the crop to a great extent, thus proving to be a major threat to the agriculture. The disease is caused by two fungal pathogens, Tilletia caries and Tilletia laevis, which are highly similar to each other in terms of life cycle and germination, but differ in morphology. This database has been implemented to provide the annotation of proteins of Tilletia species, as well as the host-pathogen interactomics tool, a platform to compare the predicted interactome of host-pathogen system.
The figure below gives an overview of the database architecture.
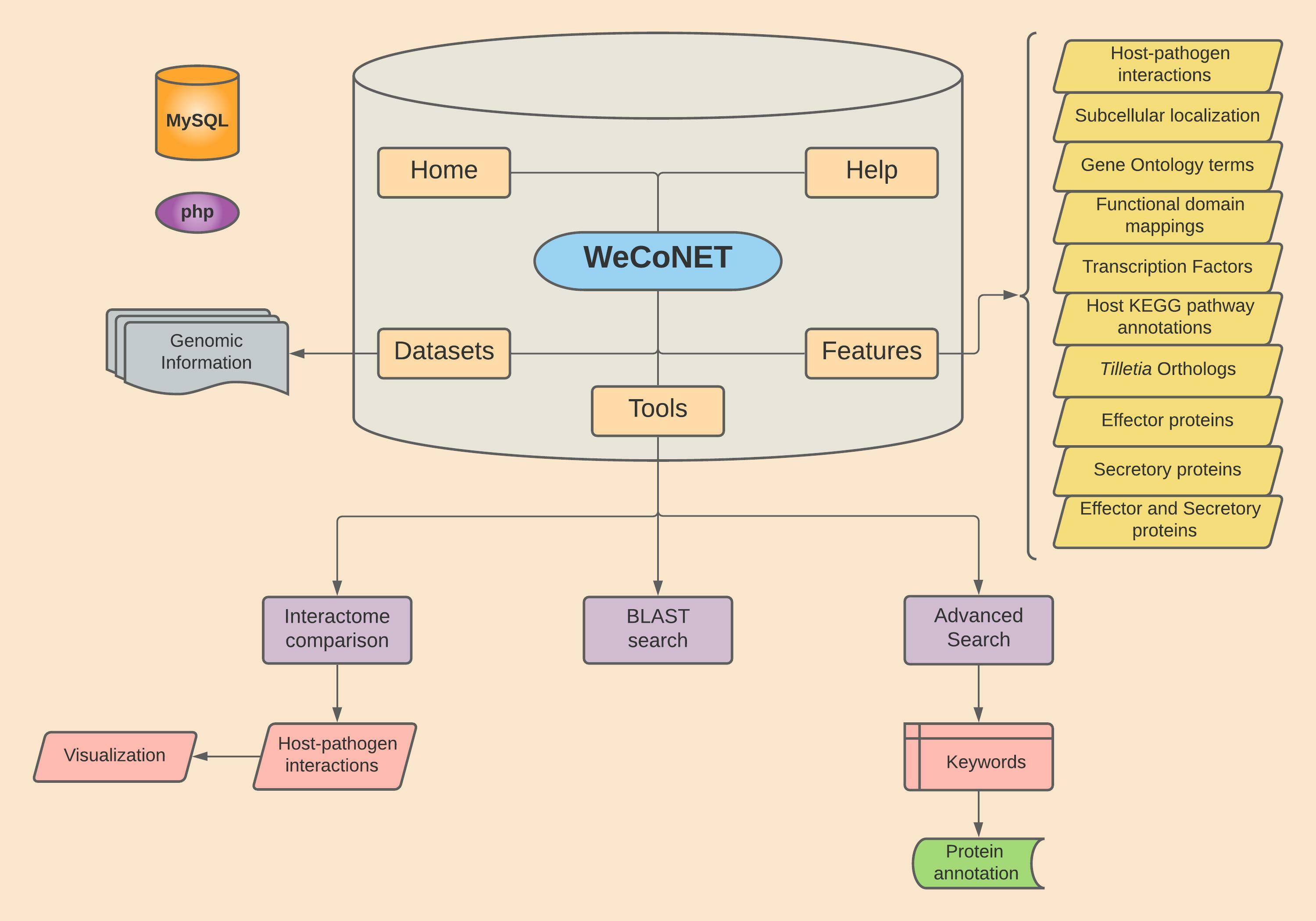
Datasets implemented in the study
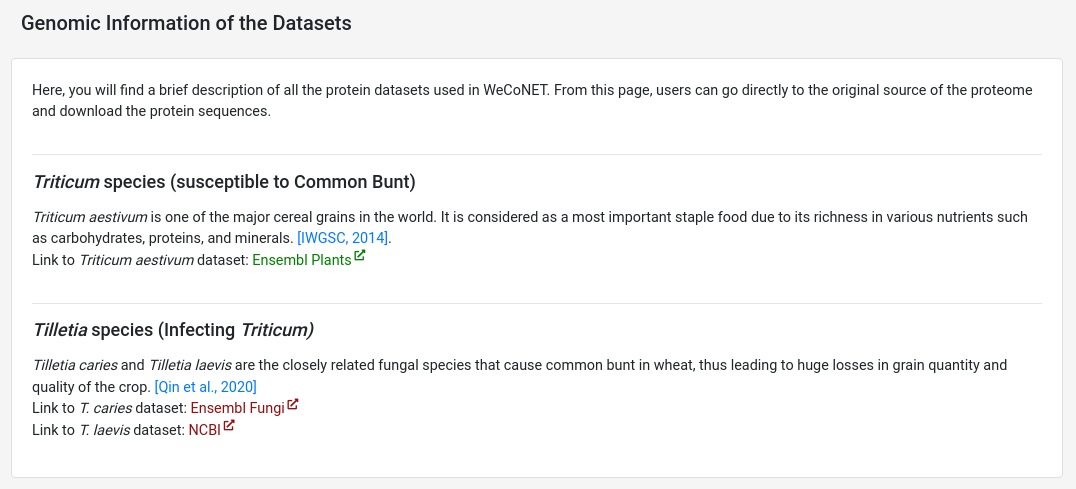
The respective host and pathogen species proteomes used in the analysis can be found on Datasets page. If required, the users can directly download the protein sequence files from these sources.
Host-pathogen interactomics module
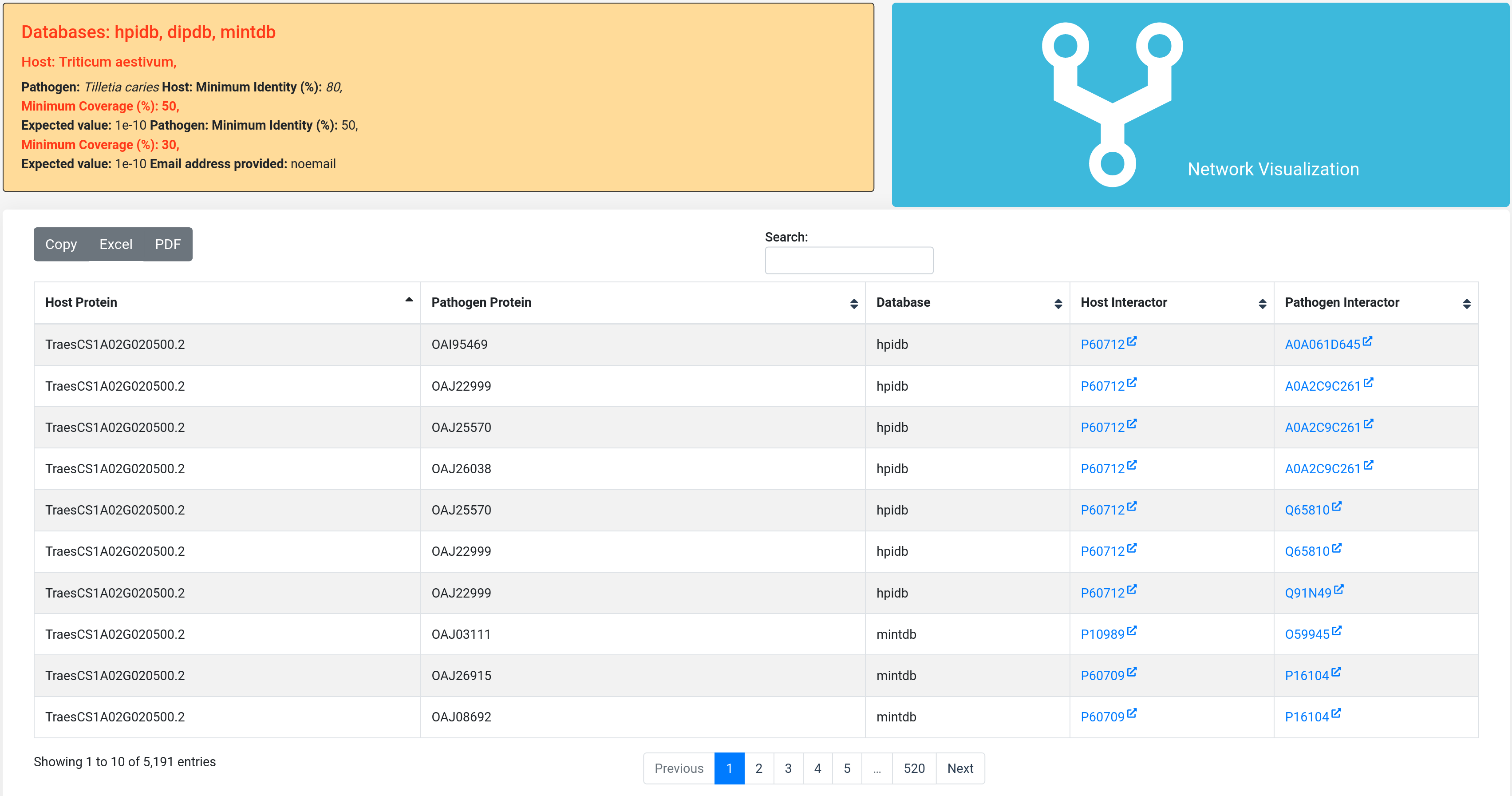
The Interactomics tool allows the user to find the interactions between host (Triticum aestivum) and pathogen (T. caries & T. laevis) proteins. In this module, the user has the option to select the specific protein-protein interaction database(s) that will be used as a template in the prediction process, or to define BLASTp alignment filters to determine homolog proteins. By default, three databases (HPIDB, MINT & DIP) have been selected. All the seven databases can be selected at once using the option 'ALL'.
The default values have been set for alignment filtering options (e-value, identity and coverage) for both host and pathogen proteins, but the tool also accepts user-entered values for these parameters.
Additionally, the user can enter the email address (optional) to receive a notification of the interactome prediction job completion.

When a job is submitted, it will be assigned to a unique identifier that user can access to check the status of the job (queried, running or done). After the job is completed, it will display the results in an enriched table with the option to sort the content by column or to be filtered by keyword. The result table can be downloaded in excel or pdf format, or copied as clipboard.
To see the respective host or pathogen protein interactor from the selected databases on the previous page, the user can click on the protein ID in 'Host Interactor' and 'Pathogen Interactor' columns, which will take the user to the respective external links. This provides the user with additional information of the specific protein. From these interactions, the user can select a specific protein of interest and search it on other features available on the database to obtain functional annotation of the particular protein.
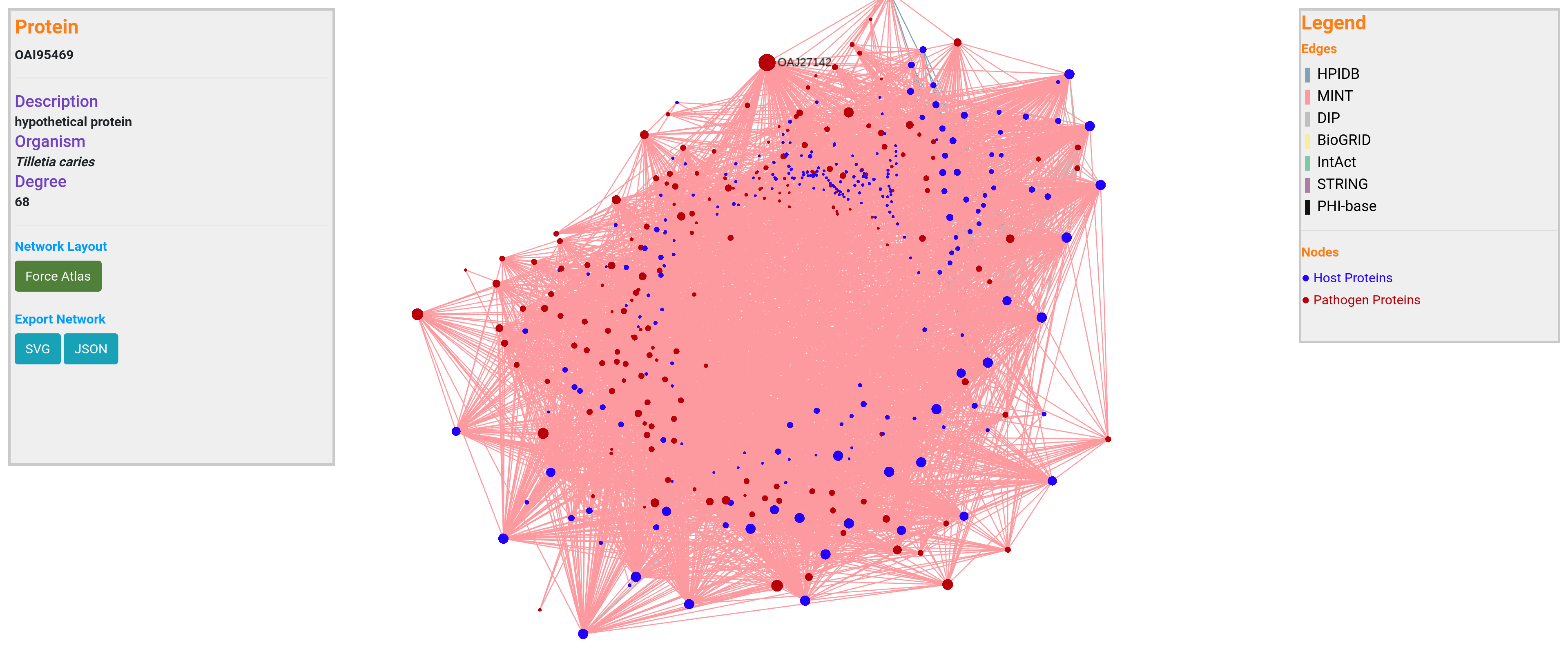
Further, the user can click on 'Network Visualization' to visualize the network of the predicted interactions.
WeCoNET provides an efficient network visualization platform, implemented using SigmaJS. This plugin was specially chosen given its performance at displaying large networks. From the host-pathogen network visualization, a user can visualize a set of traits for each node (species, description, degree), and also can easily identify hub nodes (nodes with a higher number of edges). This is useful as hub nodes have been found crucial in infectious disease pathways. A user is not limited to the network analysis that is provided through our database, the resulted network can be further examined in any network analyzer that could handle JSON or tabular network files.
In the network, the color of the edges correspond to the respective databases chosen. The edges from each database are represented with different colors as shown on the top right corner of the page. The blue nodes represent host proteins while the red nodes are pathogen proteins.
The user can click on any node (one at a time) in the network to see the respective description of the protein, which is shown on the top left corner of the page.
To analyze the network within the database, the user can select a particular node and move it around. The layout of the network can also be reset using 'Force Atlas' button.
Advanced search
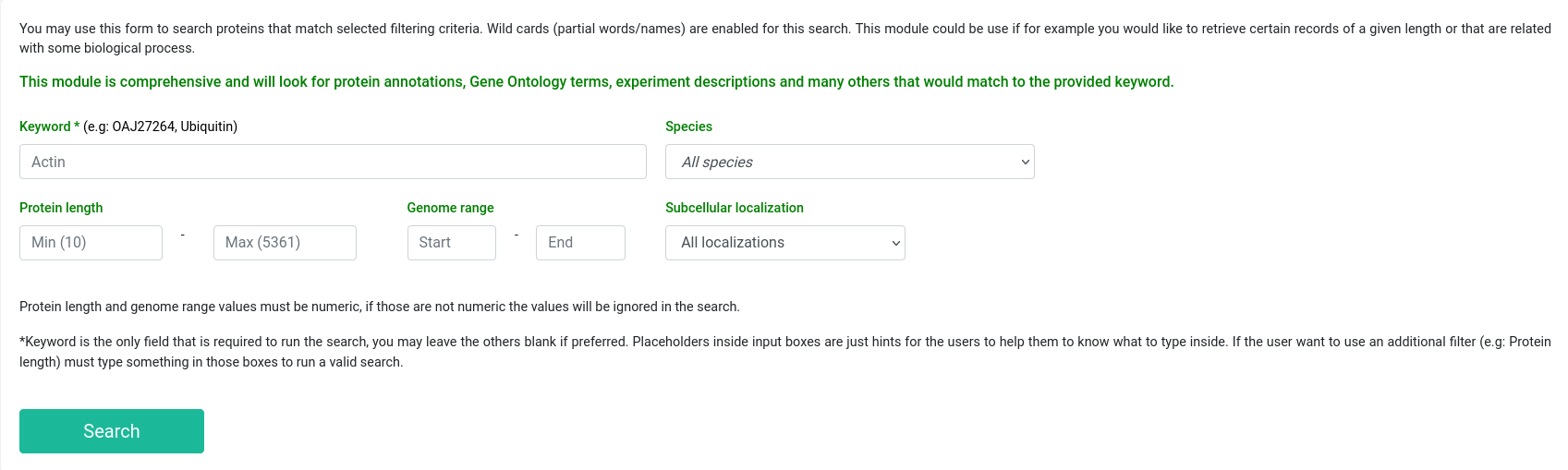
This tool provides an advanced search module that can be used to search for proteins that fulfill a selected filtering criterion, means that for a given keyword plus a set of filtering parameters, this module will look up for any record that match. This search module is comprehensive and will look for protein annotations, GO terms, experiment descriptions and many others that would match to the provided keyword. Additionally, a basic option to perform a quick search of a protein accession is available at all the pages of WeCoNET, and both the advanced and the basic search will display the complete information that can be obtained from our database records.
For example, the user can search the keyword 'actin', along with the specific subcellular localization (say 'Cytoplasm') from the dropdown menu. If there is a specific requirement for protein length or gene coordinates, the user can enter the specific value.
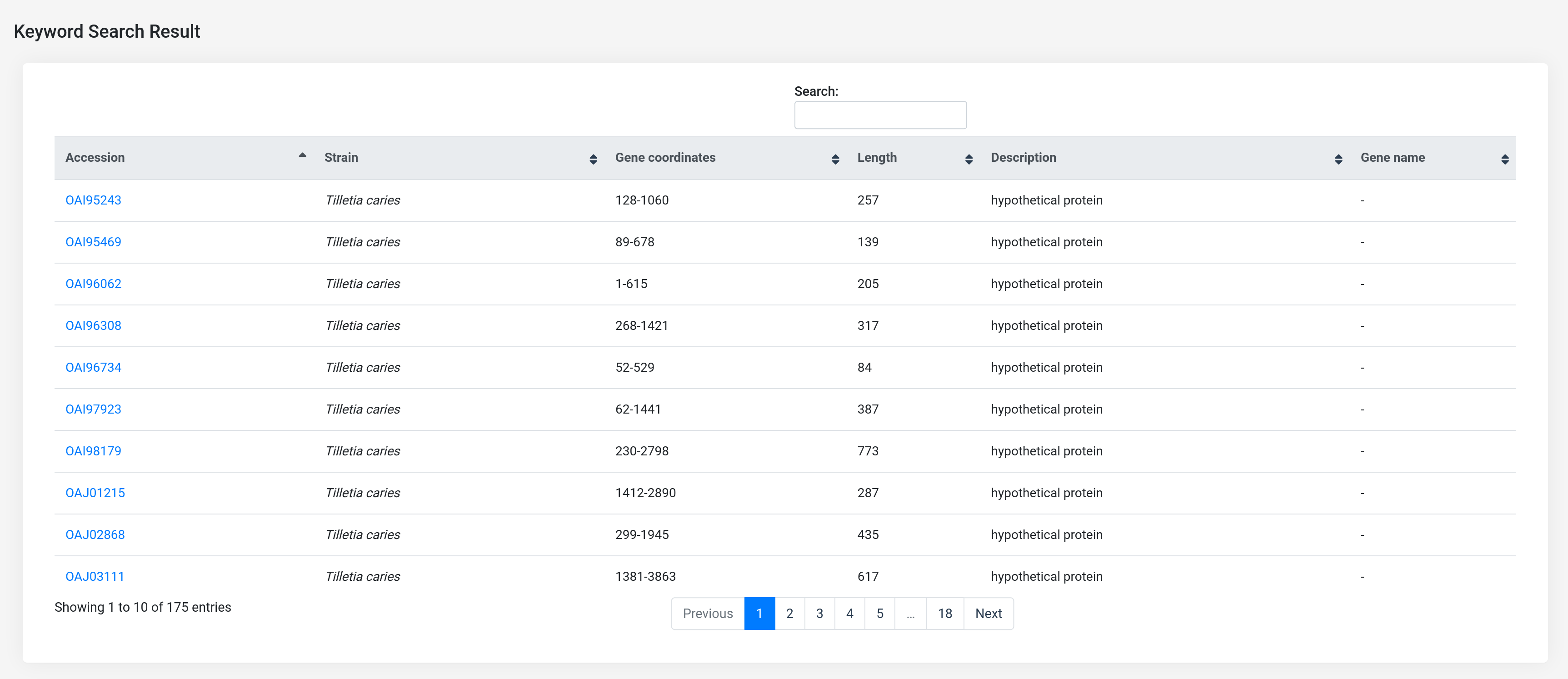
After the search is complete, the results page shown below will display a table with all the records that matched the selected filtering criteria. This contains all the proteins with the provided keyword (actin) that are located in 'cytoplasm', along with the gene coordinates, length and description of the proteins.
If the user clicks on any one of the accession links (Eg., 'OAI95243'), the user will be redirected to a new page that displays the complete information that can be obtained from our database records including protein sequence information, functional domain annotation, InterPro entry, protein length, etc. If available in the records, gene ontology of the protein will also be displayed.

BLAST search
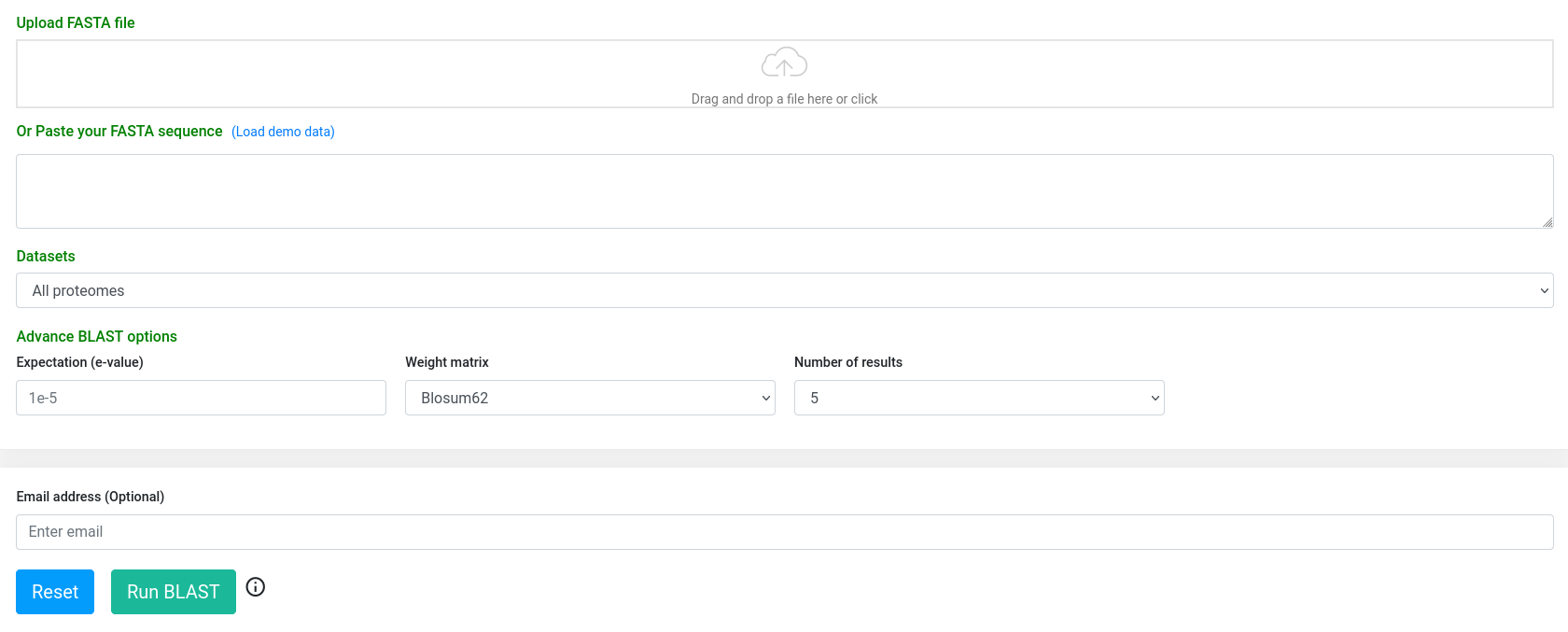
In this tool , BLAST was implemented locally in our server to provide to the user the functionality of homology sequence search. In addition to the proteome datasets, a user can select to query its sequences against 'All proteomes' (default). User can either upload a FASTA file or paste nucleotide or amino acid sequences (in FASTA format), and the system will automatically detect the specific program (BLASTp or BLASTx) to be performed.
A specific 'e-value' can be provided (default is 1e-5), and weight matrix can be selected from the available options.
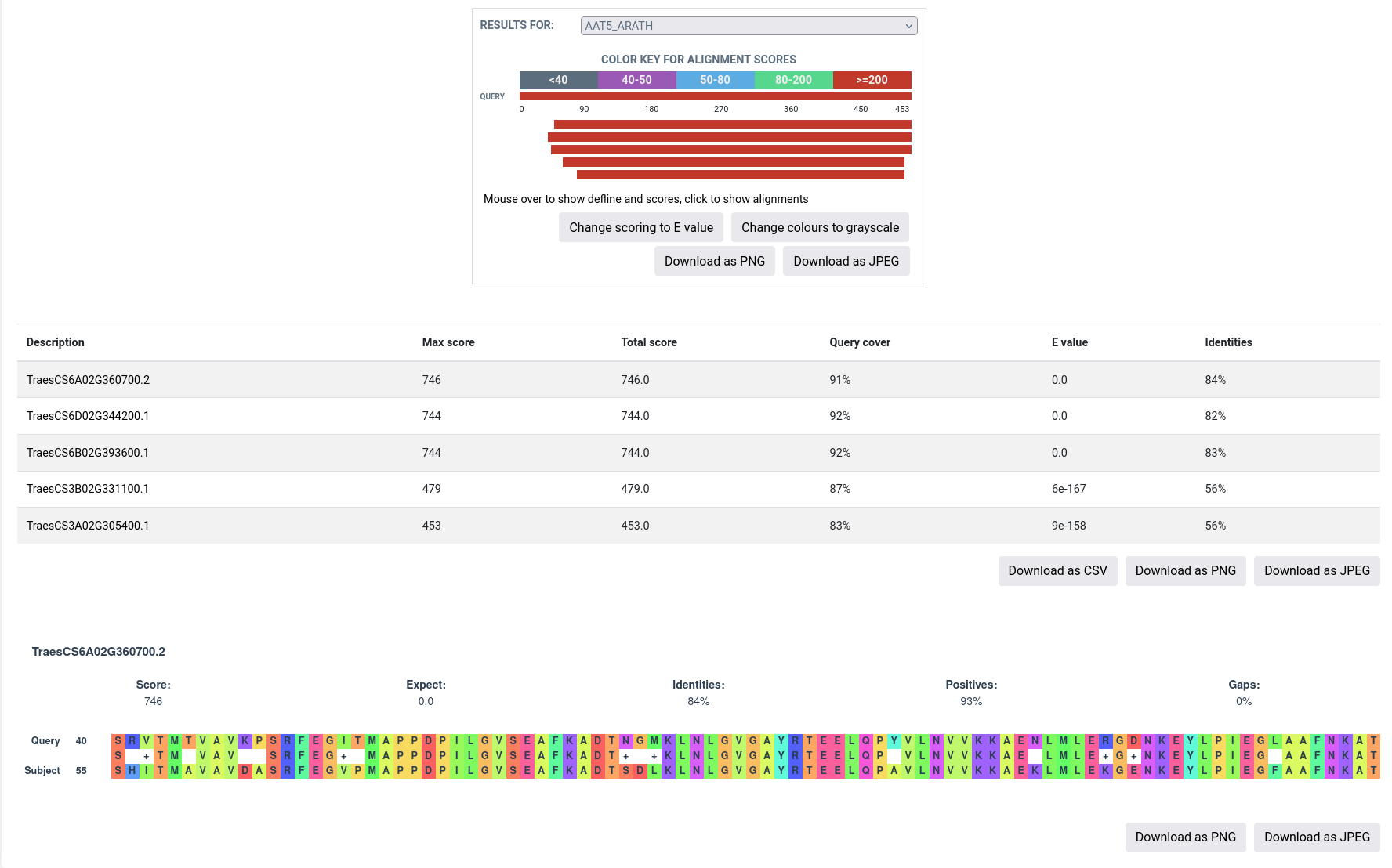
The result page (below) provides a summarized version, whereby the user can download the alignments in tabular (excel or PDF format), or standard alignment format. Also, there is a more 'detailed' option in which the alignments are visualized in an enriched mode.
Summarized

Detailed
In detailed view, the alignments for a specific protein can be viewed by selecting the protein from the dropdown menu. Further, the scoring can be changed to 'e-value' or 'max score' using "Change scoring to" button below the colored alignments. The alignments for the specific protein can also be downloaded in PNG or JPEG format for both query (top of the page) and hit (bottom of the page) obtained.
Features
Data collected from the literature or resulted from the annotation pipeline of WeCoNET is presented in different search modules for host and pathogen proteins, separately. In these modules, Tilletia protein annotations (Tilletia orthologs, Subcellular Localization Annotation, Gene Ontology (GO) Term Annotation, Functional Domain Mappings (InterPro), Effector and secretory proteins) can be retrieved. In addition, the module "Host-pathogen interactions" is available to gather interactions concerning those Triticum proteins that were found to be related with Common Bunt disease on the basis of interolog-based computational approach. The module data will be displayed according to the dataset selected.
Additionally, every feature page has an information icon that gives a brief information about the content in that particular page.
(a) Transcription factors for host : This feature includes the host proteins that serve as transcription factors along with the respective transcription factor family of the particular protein. KEGG pathway and description of the transcription facotrs has also been included on this page. Additionally, the user can search for a specific KEGG pathway (KEGG ID or description) using the search bar. The user can click on any ID (in blue) to go to the respective external links for more information about the transcription facotr family or KEGG pathway.
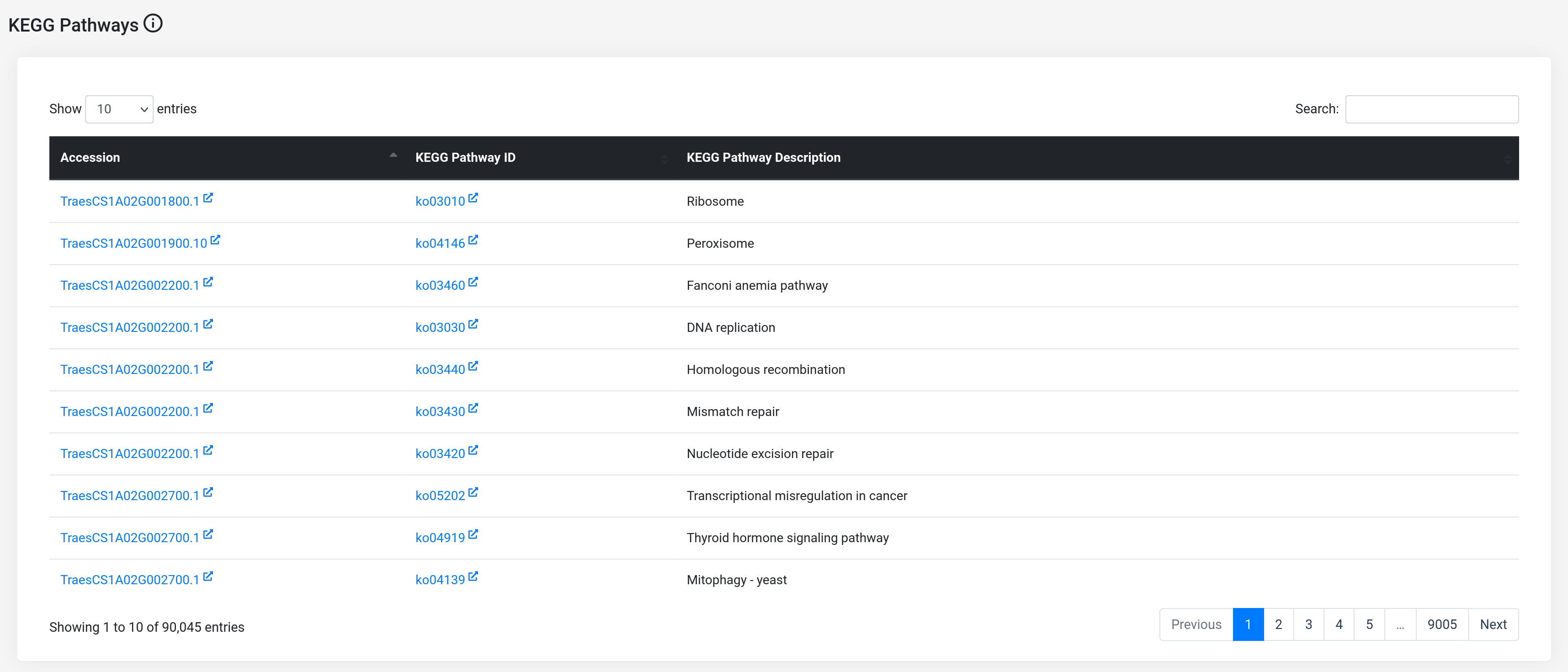
(b) KEGG pathway annotations : On this page of the database, all the host KEGG pathways and their respective description have been included. The users can further refer to the KEGG pathway website by clicking on the KEGG pathway ID.
(c) Orthologs of Tilletia species : The orthologs of T. caries and T. laevis can be found on this page. The first column i.e. 'Ortho Group' represents the unique ID of the orthologs, while the second and third columns are the T. caries and T. laevis proteins, which are the orthologs of each other. The user can click on any of the 'Ortho Group' ID to obtain the FASTA sequence of the Tilletia orthologs for that particular group.

(d) Functional domains of host and pathogen proteins : This page provides the functional domains of Tilletia and T. aestivum proteins, predicted using the InterProScan software package.

(e) Subcellular localizations of Tilletia species and T. aestivum proteins : The subcellular localizations of the host and pathogen proteins were predicted using different machine learning-based tools.
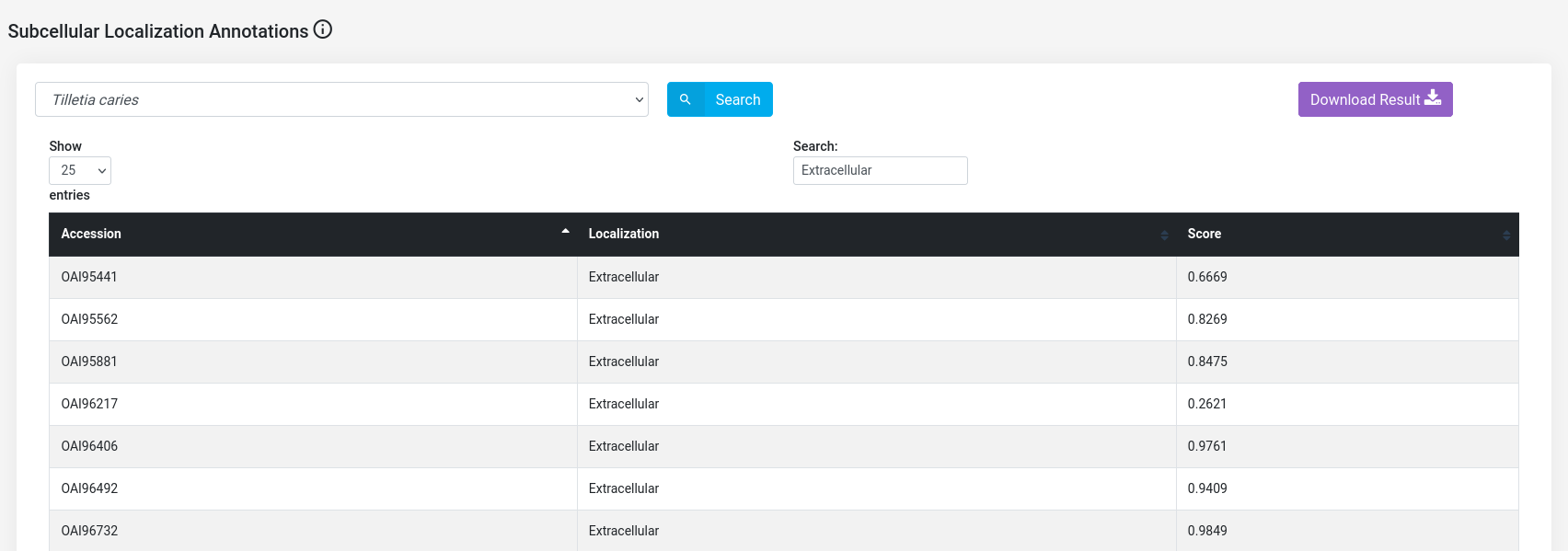
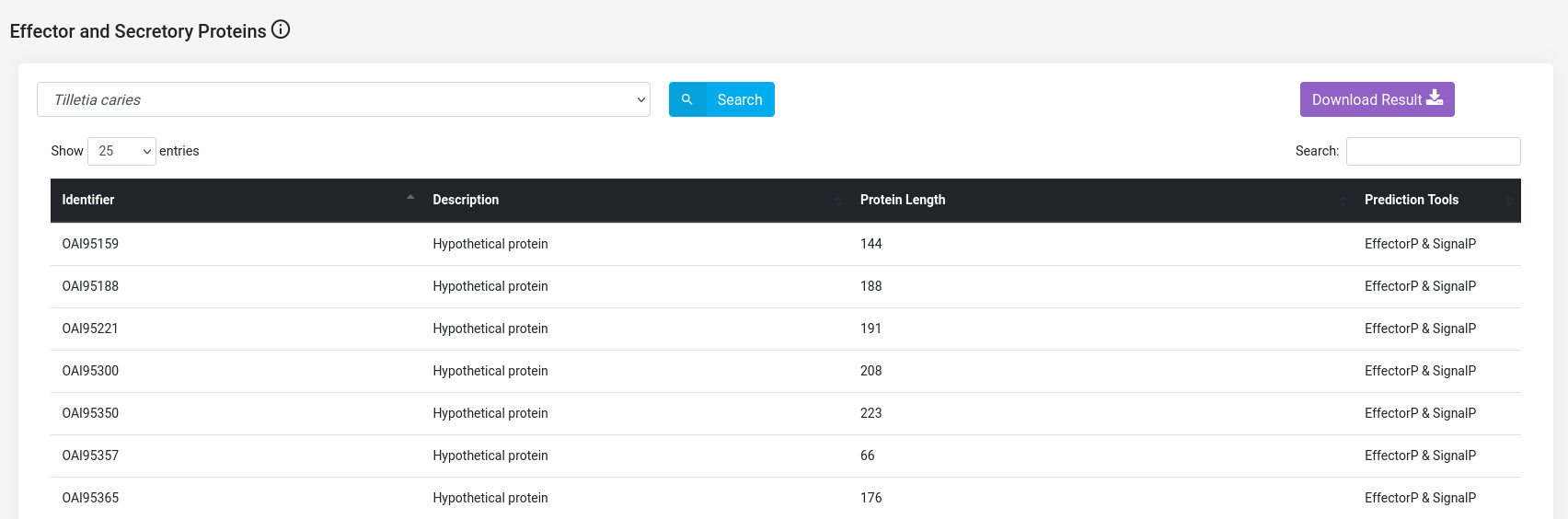
(f) Effector and Secretory proteins of Tilletia species : The T. caries and T. laevis proteins that serve as effector and secretory proteins have been separately implemented on the database. The image below shows another page that includes those Tilletia proteins that serve as both effector and secretory proteins.