| Home | Submit | How To |
- Data Submission
- Descriptor Choice
- Interpreting Data
- Amino Acid Composition
- Dipeptide Composition
- Split Amino Acid Composition
- Physicochemical Properties
- Geary Autocorrelation
- Moreau Broto Autocorrelation Descriptor
- Sequence Order Coupling Number Total
- References
Data Submission
The tool accepts a sets of fasta formatted entries
containing amino acids. Submitted amino acids must contain 50 or more residues. For some descriptors, entries may
contain small amounts of ambiguous amino acids can still be predicted. (amino acid composition, dipeptide composition,
and split amino acid composition). However, some descriptors are not able to function with make proper calculations
this these and thus they
will with a value of N/A if encountered during classification.
Descriptor Choice
Presently LigPred offers 7 different types of descriptors. Dipeptide composition has been set to the
default value as it had the highest MCC. Users may choose any combination they wish for classification.
However, it should be noted that they are
not composite descriptors but single descriptors. The predictions from each additional descriptor will be
combined with the others to make a consensus prediciton, giving higher accuracy in ambiguous cases. Below are
descriptions of the descriptors used by LigPred.
Amino Acid Composition
Geary Autocorrelation
Sequence Order Coupling Number Total
Amino Acid Composition
The fraction of each of the twenty standard amino acids in a protein sequence.
f
( r )
=
N
r
N
,
N: Length of amino acid sequence.
r = 1,2,...,20: One of the 20 standard amino acids
N
r
: Number of amino acids of type r
Dipeptide Composition
The fraction of each combination of two the twenty standard amino acids in a protein sequence.
f
(
r , s
)
=
N
rs
N-1
,
N : Length of the amino acid sequence.
r = 1,2,...,20: One of the 20 standard amino acids
N
rs
: Number of amino acids dipeptides of type r
Split Amino Acid Composition
The protein sequence is split into p parts and the amino acid composition is calculated for each part.
f
(
r
,
A 1 A 2 A n
)
=
{
A
r
A
1
,
for amino acids 1 - A 1
N
r
A
2
-
A
1
,
for amino acids (A 1
A 2
. .
. .
. .
N
r
A
n
-
A
n-1
,
for amino acids (A n-1 A n
A
n
: The position of an amino acid in the amino acid sequence.
r = 1,2,...,20: One of the 20 standard amino acids
N
r
: Number of amino acids of type r
Physicochemical Properties
A 13 element vector with each element containing a physical or chemical property of the protein.
based on the Physico-chemical property composition given on COPid
Where:
based on the Physico-chemical property composition given on COPid
Where:
element 1 = Molecular weight of the protein
element 2 = Number of amino acids in the protein sequnece
element 3 = % Composition of charged residues (DEKHR)
element 4 = % Composition of aliphatic residues (ILV)
element 5 = % Composition of Aromatic residues (FHWY)
element 6 = % Composition of Polar residues (DERKQN)
element 7 = % Composition of Neutral residues (AGHPSTY)
element 8 = % Composition of Hydrophobic residues (CVLIMFW)
element 9 = % composition of Positive charged residues (HKR)
element 10 = % Composition of Negative charged residues (DE)
element 11 = % Composition of tiny residues (ACDGST)
element 12 = % Composition of Small residues (EHILKMNPQV)
element 13 = % Composition of Large residues (FRWY)
element 2 = Number of amino acids in the protein sequnece
element 3 = % Composition of charged residues (DEKHR)
element 4 = % Composition of aliphatic residues (ILV)
element 5 = % Composition of Aromatic residues (FHWY)
element 6 = % Composition of Polar residues (DERKQN)
element 7 = % Composition of Neutral residues (AGHPSTY)
element 8 = % Composition of Hydrophobic residues (CVLIMFW)
element 9 = % composition of Positive charged residues (HKR)
element 10 = % Composition of Negative charged residues (DE)
element 11 = % Composition of tiny residues (ACDGST)
element 12 = % Composition of Small residues (EHILKMNPQV)
element 13 = % Composition of Large residues (FRWY)
Geary Autocorrelation
One of a set of topological descriptors, the Geary Autocorrelation describes the level of
correlation between a given varible and itself through space. The proterties used in this classification
system are hydrophobicity scale, average flexibility index, polarizability parameter,
free energy of solution in water, accessible surface areas, residue volume,
steric parameters and relative mutability.
With each property contributing 30 elements to create a vector with 240 elements.
Geary Autocorrelation is defined as:
f
(
d
)
=
1
2
(
N
−
d
)
∑
i
=
1
N
−
d
(
P
i
−
P
i
+
d
)
2
1
N
−
1
∑
i
=
1
N
(
P
i
−
P
¯
)
2
,
P
¯
: The average value of a property.
N : Length of the amino acid sequence.
d = 1,2,...,30: Lag of the autocorrelation.
P
i
: Property of amino acid at position i.
P
i+d
: Property of amino acid at position i+d.
Moreau Broto Autocorrelation DescriptorGeary Autocorrelation is defined as:
One of a set of topological descriptors that uses the sets of property values as the
basis for measurement. The proterties used in this classification system are hydrophobicity scale,
average flexibility index, polarizability parameter, free energy of
solution in water, accessible surface areas, residue volume, steric parameters
and relative mutability. With each property contributing 30 elements to create
a vector with 240 elements.
Moreau Broto Autocorrelation is defined as:
f
(
d
)
=
∑
i
=
1
N
−
d
P
i
P
i
+
d
,
: The average value of a property.
N : Length of the amino acid sequence.
d = 1,2,...,30: Lag of the autocorrelation.
P
i
: Property of amino acid at position i.
P
i+d
: Property of amino acid at position i+d.
Moreau Broto Autocorrelation is defined as:
Sequence Order Coupling Number Total
A 60 element vector derived from the Schneider-Wrede physicochemical distance matrix and the Grantham
chemical distance matrix between each pair of the standard amino acids.
Sequence order coupling number total is defined as:
f
(
d
)
=
∑
i
=
1
N
−
d
(
d
i
,
i
+
d
)
2
,
f(d): The dth sequence order coupling order
d: The integers 1-30.
N : Length of the amino acid sequence.
Sequence order coupling number total is defined as:
Interpreting Data
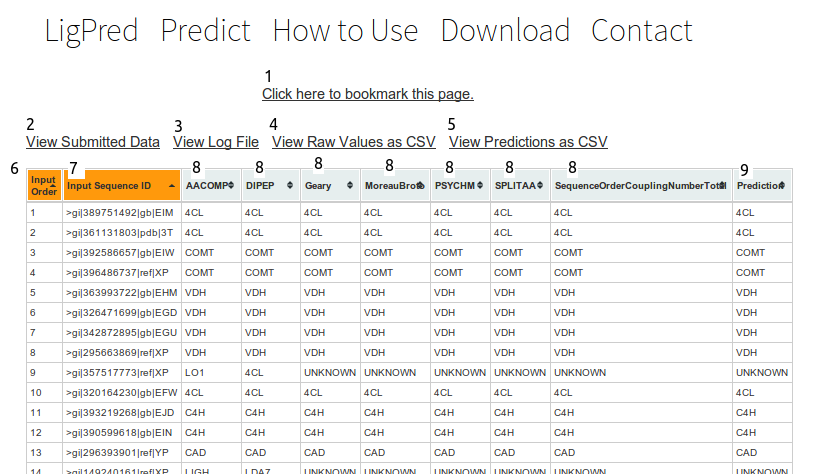
- Click this link to automaticaly bookmark the page.
- This will open a text file of the data submitted for classification.
- This will open a text file containing a log of operations preformend during classification.
- This will open a text file containing the full classification data in csv format(comma separated values) sutible for use in spreadsheets
- This will open a text file containing classification values only in csv format.
- The order fasta entries were submitted.
- The first 20 characters of the fasta entrie names
- The prediction returned by the descriptor listed in the title.
- The composite prediction.
Below is a table of the 37 classes of lignin related enzymes which can be predicted by LigPred. The classes returned by the prediction system correspond the classes in this table.
| # | Gene | Protein Name | EC# | Role | PTH | PTHP | KEGG |
|---|---|---|---|---|---|---|---|
| 1 | 4CL | 4-coumarate:CoA ligase | 6.2.1.12 | Synthesis | Uniprot | Uniprot | Kegg |
| 2 | C4H | cinnamate-4-hydroxylase | 1.14.13.11 | Synthesis | Uniprot | Uniprot | Kegg |
| 3 | CAD | cinnamyl-alcohol dehydrogenase | 1.1.1.195 | Synthesis | Uniprot | Uniprot | Kegg |
| 4 | CcoAOMT | caffeoyl-CoA O-methyltransferase | 2.1.1.104 | Synthesis | Uniprot | Uniprot | Kegg |
| 5 | CCR | cinnamoyl-CoA reductase | 1.2.1.44 | Synthesis | Uniprot | Uniprot | Kegg |
| 6 | COMT | caffeic acid 3-O-methyltransferase | 2.1.1.68 | Synthesis | Uniprot | Uniprot | Kegg |
| 7 | CS | chorismate synthase | 4.2.3.5 | Synthesis | Uniprot | Uniprot | Kegg |
| 8 | DAHPS | 3-deoxyarabinoheptulosonate-7-phosphate synthase | 2.5.1.54 | Synthesis | Uniprot | Uniprot | Kegg |
| 9 | DYP | Peroxidase DypB | 1.11.1.19 | Degradation | Uniprot | Uniprot | Kegg |
| 10 | EST | cinnamoyl esterase, glucuronoyl esterase | 3.1.1.73 | Degradation | Uniprot | Uniprot | Kegg |
| 11 | F5H | ferulate-5-hydroxylase | 1.14.-.- | Synthesis | Uniprot | Uniprot | Kegg |
| 12 | FerA | Feruloyl-CoA synthetase | 6.2.1.34 | Synthesis | Uniprot | Uniprot | Kegg |
| 13 | FerB | Feruloyl-CoA hydratase/lyase | 4.2.1.101 | Synthesis | Uniprot | Uniprot | Kegg |
| 14 | HCT | shikimate O-hydroxycinnamoyltransferase | 2.3.1.133 | Synthesis | Uniprot | Uniprot | Kegg |
| 15 | LDA1 | Aryl-alcohol oxidase | 1.1.3.7 | Degradation | Uniprot | Uniprot | Kegg |
| 16 | LDA2 | Vanillyl-aclohol oxidase | 1.1.3.38 | Degradation | Uniprot | Uniprot | Kegg |
| 17 | LDA3 | Glyoxal oxidase | 1.1.3.- | Degradation | Uniprot | Uniprot | NA |
| 18 | LDA4 | Pyranose oxidase | 1.1.3.10 | Degradation | Uniprot | Uniprot | Kegg |
| 19 | LDA5 | Glactose oxidase | 1.1.3.9 | Degradation | Uniprot | Uniprot | Kegg |
| 20 | LDA6 | Glucose Oxidase | 1.1.3.4 | Degradation | Uniprot | Uniprot | Kegg |
| 21 | LDA7 | Benzoquinone reductase | 1.6.5.(5/6/7) | Degradation | Uniprot | Uniprot | Kegg |
| 22 | LDA8 | alcohol oxidase | 1.1.3.13 | Degradation | Uniprot | Uniprot | Kegg |
| 23 | LigAB | Protocatechuate 4,5-dioxygenase | 1.13.11.8 | Degradation | Uniprot | Uniprot | Kegg |
| 24 | LigBenDiO | Lignostilbene dioxygenase | 1.13.11.43 | Degradation | Uniprot | Uniprot | Kegg |
| 25 | LigD | C alpha-dehydrogenase | 1.-.-.- | Degradation | Uniprot | Uniprot | Kegg |
| 26 | LigEFG | Beta-etherase | 2.5.1.18 | Degradation | Uniprot | Uniprot | Kegg |
| 27 | LigH | formate---tetrahydrofolate ligase | 6.3.4.3 | Degradation | Uniprot | Uniprot | Kegg |
| 28 | LigI | 2-pyrone-4,6-dicarboxylic acid hydrolase | 3.1.1.57 | Degradation | Uniprot | Uniprot | Kegg |
| 29 | LigJ | 4-oxalomesaconate hydratase | 4.2.1.83 | Degradation | Uniprot | Uniprot | Kegg |
| 30 | LigM | Vanillate/3-O-methylgallate O-demethylase | 1.14.13.82 | Degradation | Uniprot | Uniprot | Kegg |
| 31 | LO1 | Laccases | 1.10.3.2 | Degradation | Uniprot | Uniprot | Kegg |
| 32 | LO2 | Chloroperoxidase | 1.11.1.10 | Degradation | Uniprot | Uniprot | Kegg |
| 32 | LO2 | Lignin peroxidase | 1.11.1.14 | Degradation | Uniprot | Uniprot | Kegg |
| 32 | LO2 | Manganses Peroxidase | 1.11.1.13 | Degradation | Uniprot | Uniprot | Kegg |
| 32 | LO2 | Versitile Peroxidase | 1.11.1.16 | Degradation | Uniprot | Uniprot | Kegg |
| 33 | LO3 | Cellobiose dehydrogenase | 1.1.99.18 | Degradation | Uniprot | Uniprot | Kegg |
| 34 | MetF | methylenetetrahydrofolate reductase | 1.5.1.20 | Degradation | Uniprot | Uniprot | Kegg |
| 35 | PAL | phenylalanine ammonia-lyase | 4.3.1.24 | Synthesis | Uniprot | Uniprot | Kegg |
| 36 | SHDH | shikimate dehydrogenase | 1.1.1.25 | Synthesis | Uniprot | Uniprot | Kegg |
| 37 | VDH | Vanillin dehydrogenase | 1.2.1.67 | Degradation | Uniprot | Uniprot | Kegg |
References
Tang, Z. Q., Lin, H. H., Zhang, H. L., Han, L. Y., Chen, X., & Chen, Y. Z. (2007). Prediction of Functional Class of Proteins and Peptides Irrespective of Sequence Homology by Support Vector Machines. Bioinformatics and biology insights, 1, 19.
Udatha, D. B. R. K., Kouskoumvekaki, I., Olsson, L., & Panagiotou, G. (2011). The interplay of descriptor-based computational analysis with pharmacophore modeling builds the basis for a novel classification scheme for feruloyl esterases. Biotechnology advances, 29(1), 94.
Bugg, T. D., Ahmad, M., Hardiman, E. M., & Rahmanpour, R. (2011). Pathways for degradation of lignin in bacteria and fungi. Natural product reports, 28(12), 1883-1896.
Fu, L., Niu, B., Zhu, Z., Wu, S., & Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics, 28(23), 3150-3152.
Ahmad, M., Roberts, J. N., Hardiman, E. M., Singh, R., Eltis, L. D., & Bugg, T. D. (2011). Identification of DypB from Rhodococcus jostii RHA1 as a lignin peroxidase. Biochemistry, 50(23), 5096-5107.
Ong, S. A., Lin, H. H., Chen, Y. Z., Li, Z. R., & Cao, Z. (2007). Efficacy of different protein descriptors in predicting protein functional families. BMC bioinformatics, 8(1), 300.
Levasseur, A., Piumi, F., Coutinho, P. M., Rancurel, C., Asther, M., Delattre, M., ... & Record, E. (2008). FOLy: an integrated database for the classification and functional annotation of fungal oxidoreductases potentially involved in the degradation of lignin and related aromatic compounds. Fungal genetics and biology, 45(5), 638-645.
Kaundal, R., Saini, R., & Zhao, P. X. (2010). Combining machine learning and homology-based approaches to accurately predict subcellular localization in Arabidopsis. Plant physiology, 154(1), 36-54.
Tang, Z. Q., Lin, H. H., Zhang, H. L., Han, L. Y., Chen, X., & Chen, Y. Z. (2007). Prediction of Functional Class of Proteins and Peptides Irrespective of Sequence Homology by Support Vector Machines. Bioinformatics and biology insights, 1, 19.
Udatha, D. B. R. K., Kouskoumvekaki, I., Olsson, L., & Panagiotou, G. (2011). The interplay of descriptor-based computational analysis with pharmacophore modeling builds the basis for a novel classification scheme for feruloyl esterases. Biotechnology advances, 29(1), 94.
Bugg, T. D., Ahmad, M., Hardiman, E. M., & Rahmanpour, R. (2011). Pathways for degradation of lignin in bacteria and fungi. Natural product reports, 28(12), 1883-1896.
Fu, L., Niu, B., Zhu, Z., Wu, S., & Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics, 28(23), 3150-3152.
Ahmad, M., Roberts, J. N., Hardiman, E. M., Singh, R., Eltis, L. D., & Bugg, T. D. (2011). Identification of DypB from Rhodococcus jostii RHA1 as a lignin peroxidase. Biochemistry, 50(23), 5096-5107.
Ong, S. A., Lin, H. H., Chen, Y. Z., Li, Z. R., & Cao, Z. (2007). Efficacy of different protein descriptors in predicting protein functional families. BMC bioinformatics, 8(1), 300.
Levasseur, A., Piumi, F., Coutinho, P. M., Rancurel, C., Asther, M., Delattre, M., ... & Record, E. (2008). FOLy: an integrated database for the classification and functional annotation of fungal oxidoreductases potentially involved in the degradation of lignin and related aromatic compounds. Fungal genetics and biology, 45(5), 638-645.
Kaundal, R., Saini, R., & Zhao, P. X. (2010). Combining machine learning and homology-based approaches to accurately predict subcellular localization in Arabidopsis. Plant physiology, 154(1), 36-54.