
| Home | Submit | Help | Datasets |
How To Use AP-iNet
The web tool is developed to provide the interaction predictions between host and pathogen (Arabidopsis-Pseudomonas ) proteins in two separate formats: the prediction module and the search modules.
Prediction Modules
These modules are for predicting interactions in submitted sequences. There are two
types of prediction models avalible for each submission type: Support vector machine
(SVM) and homology based.
Support Vector Machine (SVM) Prediction
Support vector machine is a type of supervised learning algorithm and has
been widely used in bioinformatics for a variety of applications. AP-iNET used
the SVMlight implementation through the
python wrapper pysvmlight.
In AP-iNET, it take the input sequences provided by the user and derive the feature vectors.
Then this feature vectors are used to predict the interaction by the SVM models developed from the known
interaction datasets(See datasets for more details).The various features used in the SVM model is described below:
- Amino Acid Compsition (AAC): Each protein is defined by a 20-dimensional
feature vector in Euclidean space. The protein corresponds to a point whose
co-ordinates are given by the occurrence frequencies of the 20 constituent amino
acids.Each pair host-pathogen PPI is represented by a 40 length feature vector
by combing their individual amino acid composition.

-
Dipeptide Composition (DIPEP): It counts the occurrence frequencies of each dipeptide
(pair of amino acids,exp:“AA”, “AC”, “AD”...etc).This produce a fixed pattern of length 400
(20x20) for each protein and each pair of host-pathogen is represented by a 800 length feature vector.

- Conjoint triad (CT): First, the twenty native amino acids are grouped into seven classes based on their electrostatic and hydrophobic properties such as dipoles and volumes of the side chains [Shen et al.,2007]. It considers one amino acid and its vicinal amino acids and regarded any three continuous amino acids as a unit called triad. For each protein, the frequency of each triad occurred in the sequence are counted and projected in a vector space by normalizing between 0 and 1. Thus, the protein is represented by a 343-dimension (7x7x7) feature vector. Thus, the CT descriptors of the host and pathogen proteins are concatenated and a total 686-dimensional vector constructed to represent each PPI.
- Composition-Transition-Distribution (CTD):In this representation three local descriptors, Composition (C), Transition (T) and Distribution (D) are used in combination to construct the feature vector. These descriptors are based on the variation of occurrence of functional groups of amino acids within the primary sequence of protein. Thus, before computing this feature the twenty amino acids are clustered into seven functional groups based on the dipoles and volumes of the side chains [Shen et al.,2007]. The composition descriptor computes the occurrence of each amino acid group along the sequence. Transition represents the percentage frequency with which amino acid in one group is followed by amino acid in another group. The distribution feature reflects the dispersion pattern along the entire sequence by measuring the location of the first, 25%, 50%, 75% and 100% of residues of a given group. Hence, total 63 features (7 composition, 21 transition and 35 distribution) are constructed to represent a protein. Finally, the CTD descriptor of the host and pathogen is concatenated to form a 126 feature vector for each protein pair.
Homology Prediction
Homology based model relies in protein seuence similarity to conduct the protein-protein interaction prediction.
A protein pair is predicted to interact with each other if an experimentally evidenced interaction exists between their respective homologous proteins in another organism.
The homologous interacting pairs are searched against a well curated experimental evidenced interaction database [HPIDB+APINET database].
The HPIDB database (http://www.agbase.msstate.edu/hpi/main.html) is a database of experimental determined interactions between 62 hosts and 529 pathogens with 23735 unique PPIs.
The APINET database is a collection of 166 expeimentally proved PPIs between Arabidopsis-Pseudomonas from Mukhtar's expeiment (Mukhtar et al. 2011) and several other public
databses such as BIND, APID, iRefIndex, IntAct and Uniprot.
Each protein is BLASTed against the set of known PPIs in the HPIDB+APINET database with an user defined E-value cutoff and bitscore value. For each protein pair, the interaction is predicted if their corresponding homologs in the curated database have atleast one interaction.
Host-Pathogen Prediction
For a set of host proteins as well as pathogen proteins,
predict the interaction between them. Note: Host and pathogen sequence should be
submitted in their designated area as separate files or inputed in separate text areas.
There are two types of prediction avalible a support vector machine based model and a
homology model. The model used for prediciton can be decided by setting the radio button
under "Prediction Method" to the desired method.
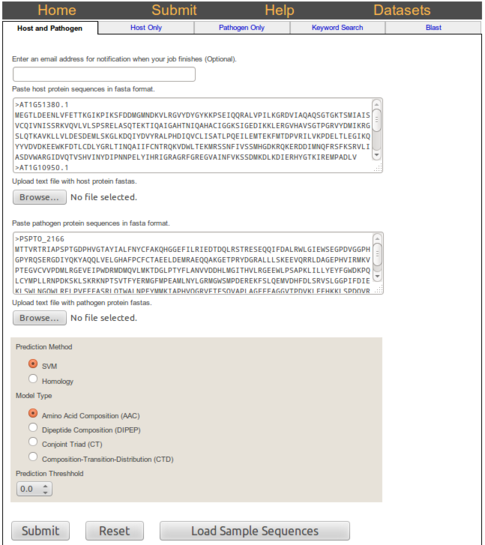
When the submission finishes a results page will be shown. The results page contains a table in which
each row shows the score returned by the SVM prediction for a host-pathogen protein pair and the descision
based on the cutoff value supplied. Additionally, the links at the top show a link to a cytoscape graph of the
interactiosn between the proteins submitted as well as offer links to download the data submitted
as comma separated values (CSV), tab separated values (TSV)
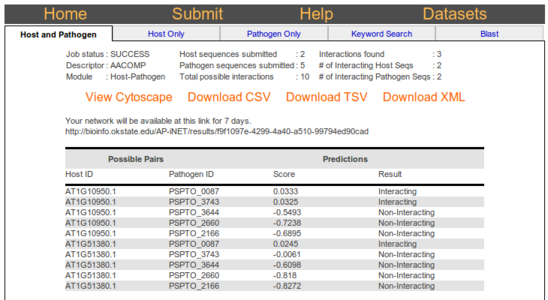
Clicking on the cytoscape link will open a cytoscape viewer. In the cytoscape viewer the interactions are
represented as lines between colored nodes which represent proteins. Clicking on a node will display
additional information about the node in the box below the graph.
Host Interaction Predction
For a set of host proteins, predict their interaction with specific strains of
Pseudomonas syringae. Note: Host sequences should be inputed in
their designated text area or file upload.
Pathogen Prediction
For a set of patghogen proteins. predict their interaction with Arabidopsis.
Note: Pathogen sequences should be inputed in their designated text area or file upload.
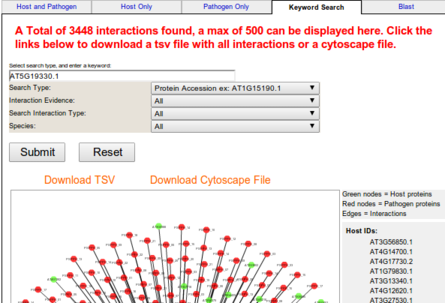
Keyword Search
This provides the facility that user can provide any specific function (GO term, domain name, etc.) or
an accession number of host or pathogen as a query to see its interacting partners with their functional descriptions.
The keyword search is a tool for searching the AP-iNET database consiting of all
experimentally validated interactions usd in AP-iNET as well as the predicitons made by
AP-iNET between the strains of Psudomonas and Arabadposis.

Search queries will return sequences matches by the given query and sequences
that interact with the matched sequences.

Blast Search
The "Blast" tab allows users to search the AP-iNET database for interactions between sequences
homologous to the query.

The results show the e-value an bitscore between the query and the hit. Clicking
on the "View Cytoscape" link will open a search with the
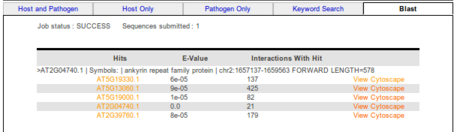
The search opened functions identically to searching an accession number in "Keyword Search" tab.
Inputting Sequences
Please enter the sequnces into the designated areas for host or pathogen! This can directly affect the accuracy of the prediction! You can either copy/type sequences in the box or upload a file. Max input is noted in upload area. Please input FASTA formatted sequence only.
FASTA format example:
< Seq1
MASTPGHTIIYEAVCLHNDRTTIPMANASGFFTHPSIPNLRSRIHVPVRVSGSG
>Seq2 optional comment
MASQKRPSQRHGSKYLATASTMDHARHGFLPRHRDTGILDSIGRFFGGDRGAPK
NMYKDSHHPARTAHYGSLPQKSHGRTQDENPVVHFFKNIVTPRTPPPSQGKGRV
KSAHKGFKGVDAQGTLSKIFKLGGRDSRSGSPMARRELVISLIVES
Enter your email (optional)
Results may take a few minutes depending on the amount of sequences uploaded. A link to a generated page for your results will be sent to your supplied email address. This link will be viewable for a set amount of time (7 days). This is optional. A link to results will also be supplied on a redirected page after sumission.
Submit or Clear
When ready hit submit to start the prediction.
Interpretion of SVM results
The prediction results shows sequence IDs with the SVM score. The score greater than 0.00 are considered positive for being interacting proteins in a host-pathogen network and scores below 0.00 are considered non-interacting proteins in a host-pathogen network.